Alanine-scanning mutagenesis has proven to be a key experimental tool to identify the residues that are most important for mediating interactions between a protein and a ligand (which could be a small molecule, a nucleic acid, a different protein, or a different region within the same protein, etc.). However, mutating individual / combinations of amino acids to alanine experimentally is both time-consuming and costly. Consequently, several computational alanine scanning (CAS) programs have been developed that enable users to identify hotspot residues that make the greatest contribution to the interaction of interest in silico.
In two recent papers [1, 2], the Sessions and Woolfson groups at the University of Bristol, in collaboration with the Wilson group at the University of Leeds, introduce BUDE Alanine Scan (BAlaS), a command-line tool to run CAS within the BUDE force field [3, 4]. BAlaS enables high-throughput CAS of both individual X-ray crystal structures plus ensembles of structures obtained from NMR / MD analysis. In Ibarra et al. (2019), the authors introduce the BAlaS software, and benchmark its performance relative to a range of pre-existing CAS software, firstly on the SKEMPI database (a database of 3047 binding free energy changes upon mutation, collated from the literature, for PPIs with known structure), and secondly on newly acquired alanine-scanning mutagenesis data collected for three diverse protein-protein interactions. They find that BAlaS predicts the experimentally identified hotspot residues with comparable accuracy to the best-performing CAS software, whilst running considerably faster. However, the authors also find that averaging the results from all CAS software tested achieves better results than any of the individual programs, and hence recommend CAS users adopt such a meta-scoring approach.
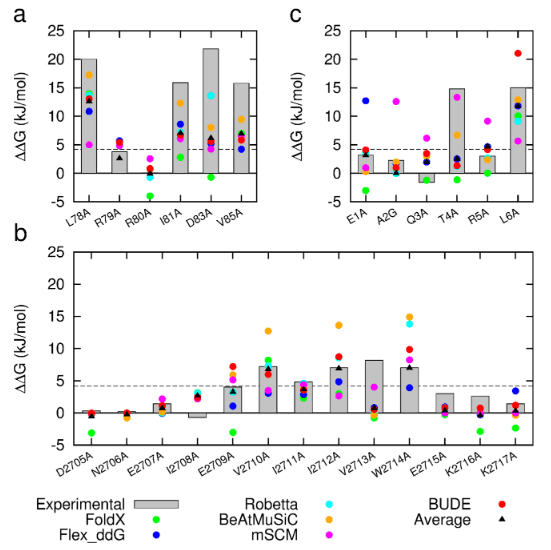
Figure 1: Comparison of experimentally measured vs. computationally predicted (via 6 different CAS software packages) DDG values for individual alanine mutations of interface residues in three diverse protein-protein interactions: a) NOXA-B/MCL-1 (PDB model 1); b) SIMS/SUMO (PDB model 1); and c) GKAP/SHANK-PDZ (chains A and C).
In Wood et al. (2020), the authors introduce an interactive web application to run BAlaS (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). This interface is simple and intuitive, allowing non-expert users to easily run CAS on their own protein-ligand interactions of interest. In addition to allowing mutation of individual residues to alanine, the interface also allows the user to simultaneously mutate constellations of residues (selected either manually or via all permutations of a specified constellation size within a selected subset of residues) to alanine in order to identify “hotspot patches” of residues within the ligand. The capability to perform mutations to residues other than alanine, available in the command-line tool, will be introduced in a future version of the web application.
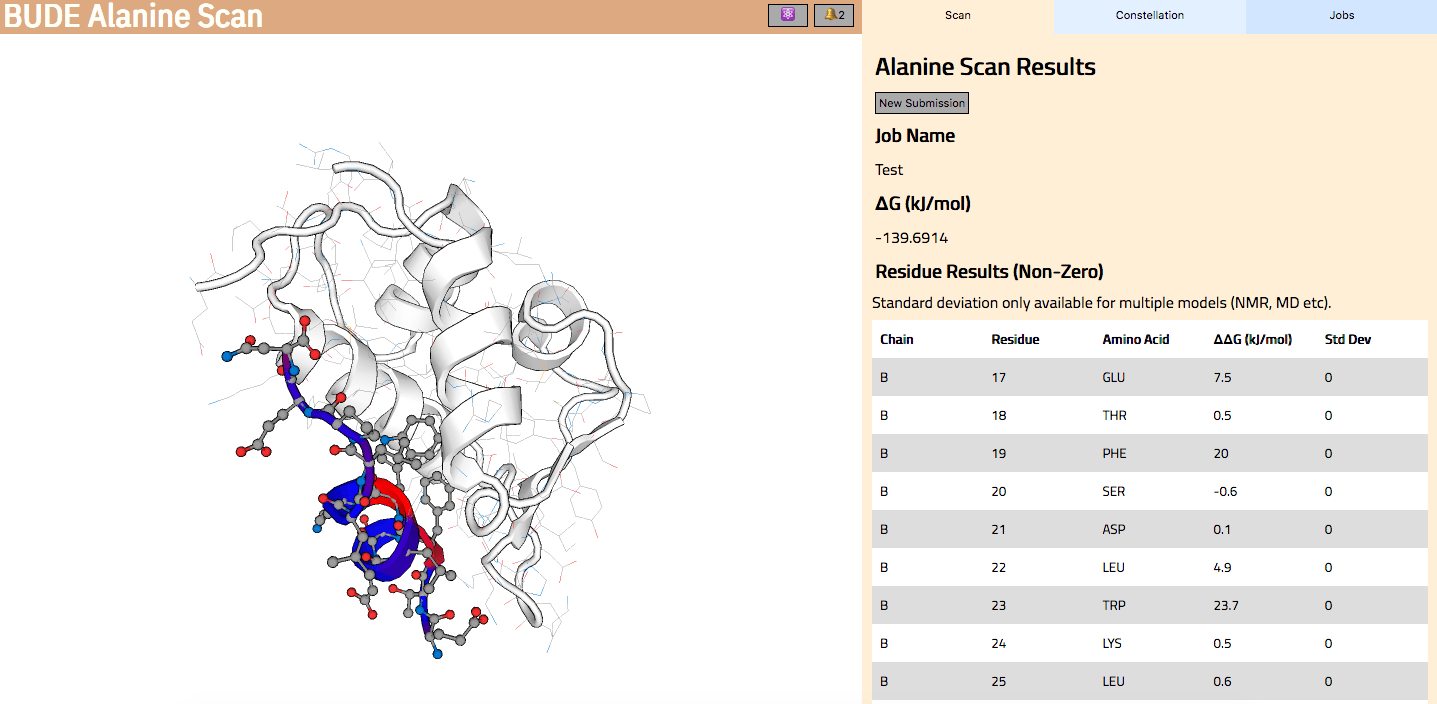
Figure 2: The BAlaS web interface. Here DDG values are calculated for residues in the transactivation domain of p53 (the “ligand”) bound to MDM2 (the “receptor”) (PDB structure 1YCR).
References
- A. A. Ibarra et al., Predicting and experimentally validating hot-spot residues at protein–protein interfaces. ACS Chem Biol 14, 2252-2263 (2019).
- C. W. Wood et al., BAlaS: fast, interactive and accessible computational alanine-scanning using BudeAlaScan. Bioinformatics (2020).
- S. McIntosh-Smith, T. Wilson, A. Á. Ibarra, J. Crisp, R. B. Sessions, Benchmarking energy efficiency, power costs and carbon emissions on heterogeneous systems. The Computer Journal 55, 192-205 (2012).
- S. McIntosh-Smith, J. Price, R. B. Sessions, A. A. Ibarra, High performance in silico virtual drug screening on many-core processors. The International Journal of High Performance Computing Applications 29, 119-134 (2015).