Intricate molecular choreography separates the bound and unbound states of proteins, resulting in slow binding kinetics that make simulating these events difficult. Interactive molecular dynamics (iMD) allows simulations to be driven to a desired state using a combination of human chemical and spatial intuition and current advances present virtual reality (VR) as a novel strategy for simulation biasing. Previous work shows VR interfaces to iMD (iMD-VR) are sufficiently performant for basic molecular manipulation tasks, but this begs the question of whether these interfaces can be used for more complex tasks, namely correctly establishing bound poses between proteins and ligands.
Our iMD-VR framework for interactively chaperoning drug unbinding and rebinding events was tested on a cohort of non-expert users (n=10), defined as being unfamiliar with the iMD-VR interface. Three example protein-ligand systems were chosen, and participants were asked to undock and redock a ligand from these proteins twice, first with the aid of a trace representation of the ligand in the correct position to guide them, and once again with no trace guide present. Below is a representation of two people in VR undocking a ligand from a protein, and also the three protein-ligand systems used in the study, with key interactions highlighted.
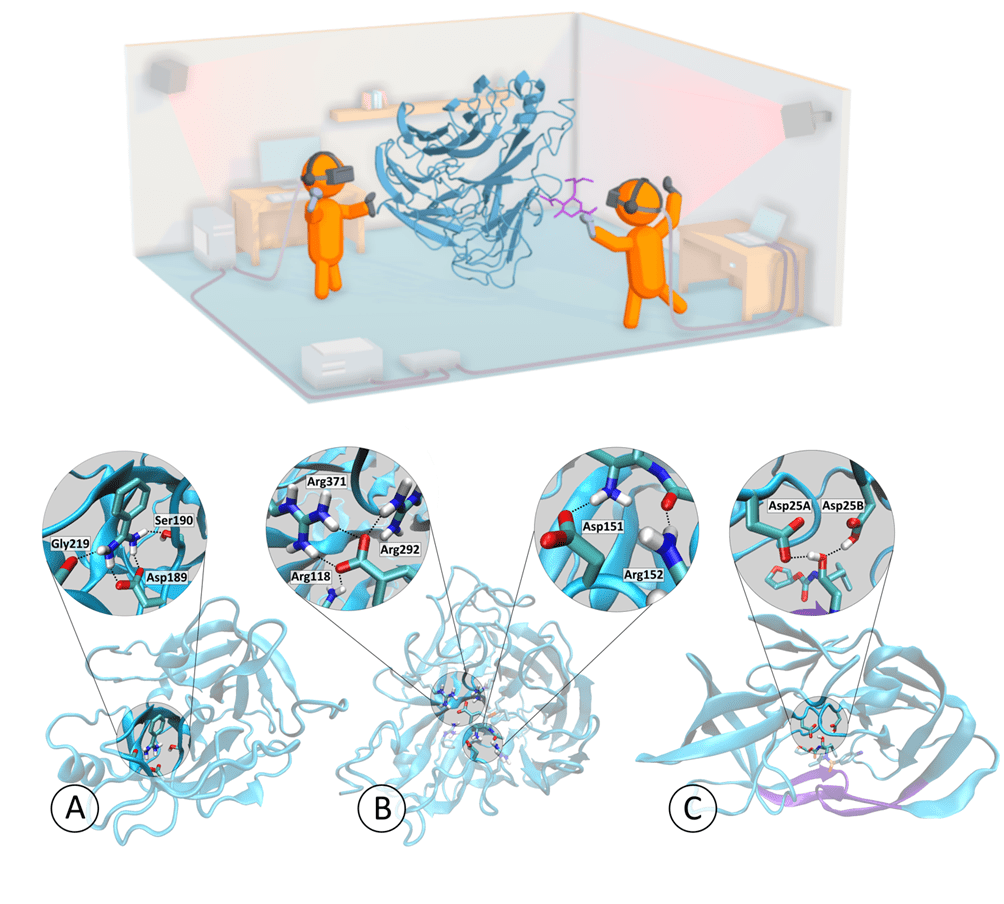
We show that it is possible to unbind and rebind ligands from three protein targets in a simulation timescale totalling less than 50ps, simulated at a rate of 4.5 ps/min of real time. Where non-expert users had trace atoms showing them the correct pose, all users were able to establish a docking pose within 2Å of the starting structure (1Å for two out of the three tasks). Where no trace atoms were present, binding poses understandably had higher variation, however, participants were still able to get within the same range of RMSD for all three systems. These results were achieved within a single hour-long training session with each participant and are presented below.
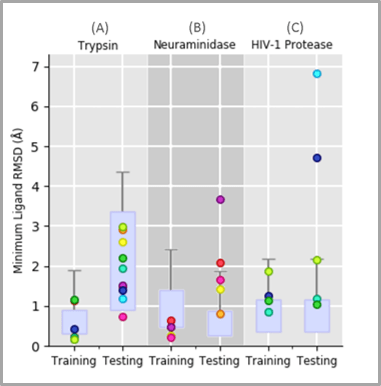
Our results show that, even with very limited experience, iMD-VR is performant enough to allow (a) unbinding of a ligand from a protein binding pocket and (b) re-establishment of the original binding pose, demonstrating iMD-VR as a useful tool for sampling states where a ligand is both bound and unbound from a protein.
Helen M. Deeks, Rebecca K. Walters, Stephanie R. Hare, Michael B. O’Connor, Adrian J. Mulholland and David R. Glowacki
PLOSONE, 2020
doi: to be added very soon!