Constant pH Molecular Dynamics (CpHMD) is a simulation technique gaining in popularity owing to its ability to titrate ionisable residues and predict pKa’s using standard molecular mechanics force fields. State of the art CpHMD simulations involve running pH replica exchange MD (pH-REMD) simulations using an explicit solvent model to accurately sample the protein conformational and protonation state spaces. It has been successfully applied to a variety of natural proteins to estimate the pKa of key residues and provide mechanistic insights into pH-dependant phenomena.
In a recent paper published in Nature Communications, BCompB members Eric Lang and Adrian Mulholland pushed the boundaries of CpHMD simulations further by investigating the pH dependency of de novo α-helical barrels designed with a ring of 6 or 7 of strongly interacting glutamate residues whose side chains point toward the inside of the central, otherwise hydrophobic, lumen.
The CpHMD simulations were used to investigate the pH-dependant stability of the hexameric and heptameric α-helical barrels (named CC-Type2-LL-L17E and CC-Type2-IL-Sg-L17E respectively) observed experimentally by circular dichroism (CD). For each barrel, two 200 ns pH-REMD simulations in TIP3P water, were carried out with all ionisable residues allowed to titrate. A total of 16 replicas was used to cover a pH range between 3.0 and 10.5, with one replica per 0.5 pH unit, representing an aggregate sampling time of 6.4 µs for each structure.
Because the glutamate residues (Glu-17) are in close proximity and interact strongly, the authors anticipated that the individual microscopic pKa of each glutamate residue made little sense and instead considered the Glu-17 rings as single polyprotic species composed of 6 or 7 titrable groups. Using this approach, a macroscopic stepwise pKa can be calculated for the (Glu-17)6 and (Glu-17)7 species, giving results in excellent agreement with the experimental data. These calculations revealed that up to two negative charges can be accommodated inside the barrels but increasing the number of charged Glu dramatically decreases the stability of the barrels. The simulations also revealed that the two buried negative charges are accommodated due to the solvation of the rings of Glu-17 residues and the presence of a Na+ counter ion in the middle of the rings. Increasing the number of deprotonated Glu-17 above two leads to a reorientation of the Glu side chains to access bulk solvent. This, in turn, leads to disruption of the secondary structure and ultimately to the opening of the barrels.
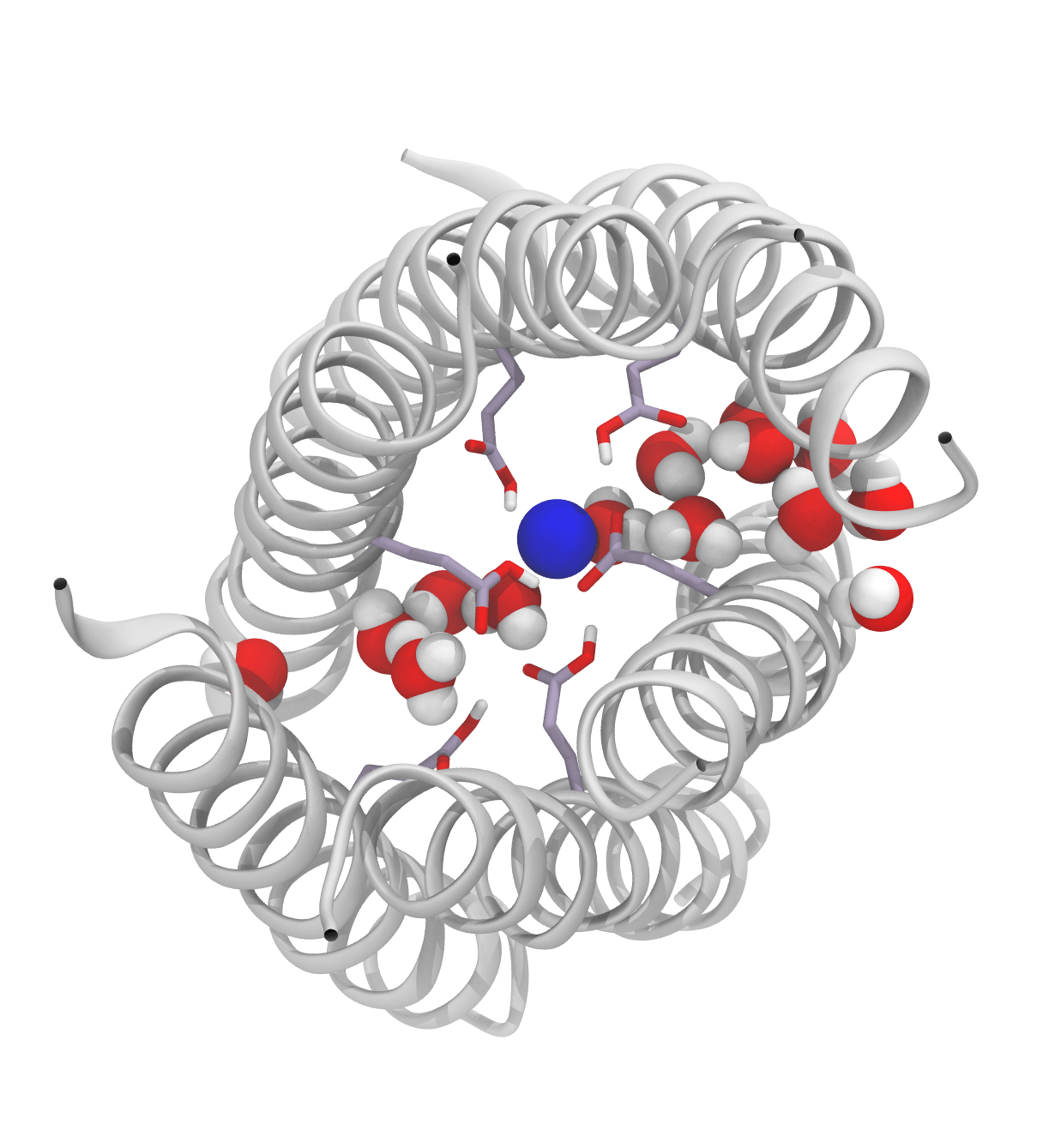
The ring of Glu-17 residues in the central lumen of a hexameric α-helical barrel
The paper, which primarily presents the experimental and design work lead by Guto Rhys and Derek Woolfson, also provides an example of how the Isambard software, developed in the Wolfson group, was used by BCompB member Christopher Wood to rationalise the diversity of structures formed by the peptides studied in this work.