In this recent publication included in the ‘Women in Computational Chemistry’ special issue of the Journal of Chemical Information and Modeling, we investigate the dependence of predicted reaction energetics on the choice of density functional and QM region size used in QM/MM calculations. DFT methods potentially offer a good combination of accuracy and computational cost but suffer from some well-known limitations and are not systematically improvable. The Claisen rearrangement of chorismate to prephenate, a simple unimolecular reaction catalysed by the enzyme chorismate mutase, was used as a model system to investigate how these choices for QM/MM calculations impact the predicted energetics of reaction.
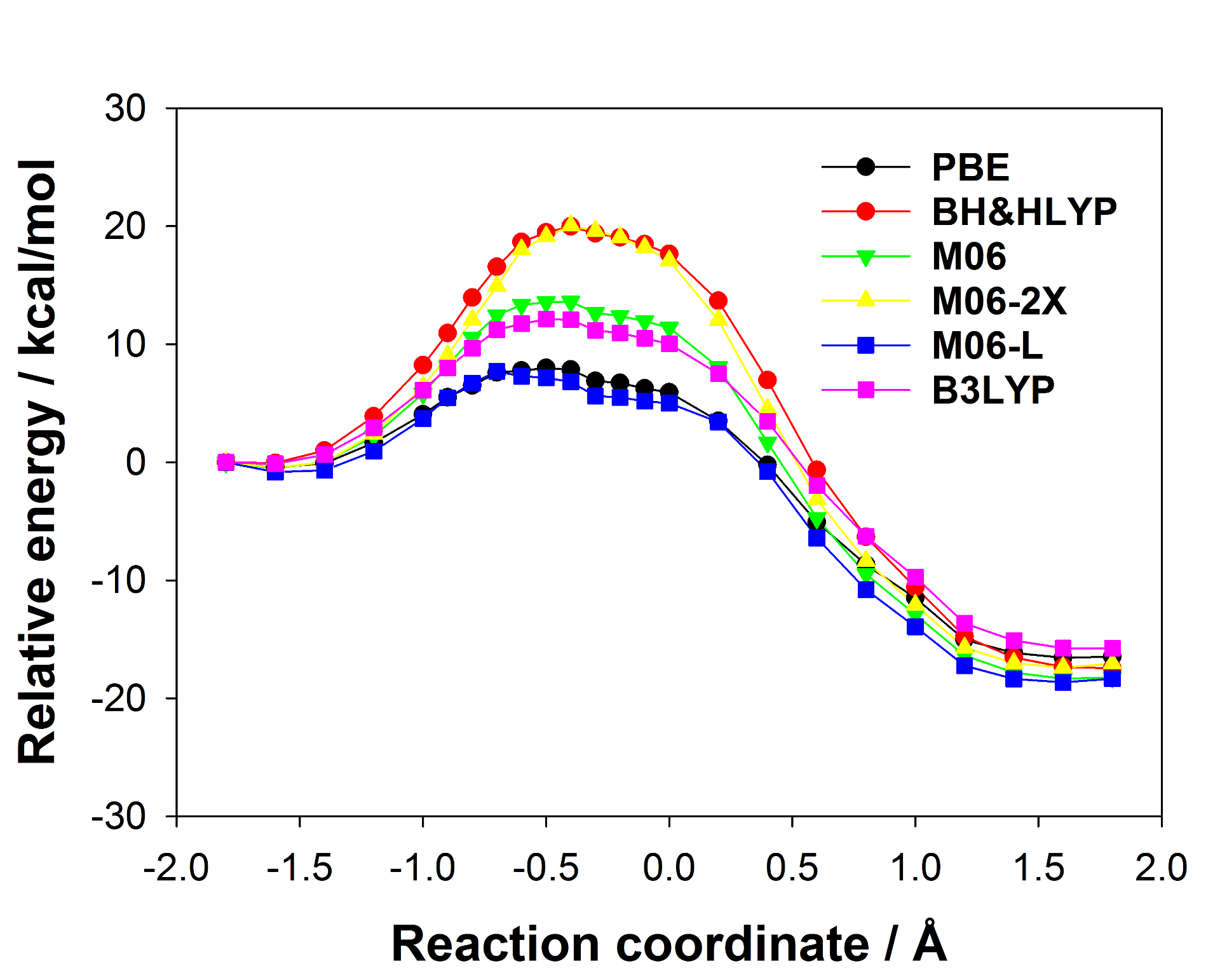
There are many different density functionals available and the best density functional to choose for any application is not obvious. Commonly used density functionals (e.g. B3LYP, BH&HLYP, MO6-2X) predict barrier heights for the reaction in the enzyme that differ by as much 13 kcal/mol. When different density functionals give different results for the same system, which result should be preferred? Ab initio methods are potentially highly accurate and are systematically improvable but often restricted by their computational cost. Projector-based embedding allows the incorporation of ab initio methods into QM/MM calculations at reasonable computational cost as a small number of atoms can be selected for treatment at the highest levels. Embedding SCS-MP2 in DFT in these QM/MM calculations reduced the spread in predicted barrier height from 13 kcal/mol to 0.3 kcal/mol, essentially eliminating the dependence on the density functional, as shown below (top: standard DFT, bottom: SCS-MP2-in-DFT).

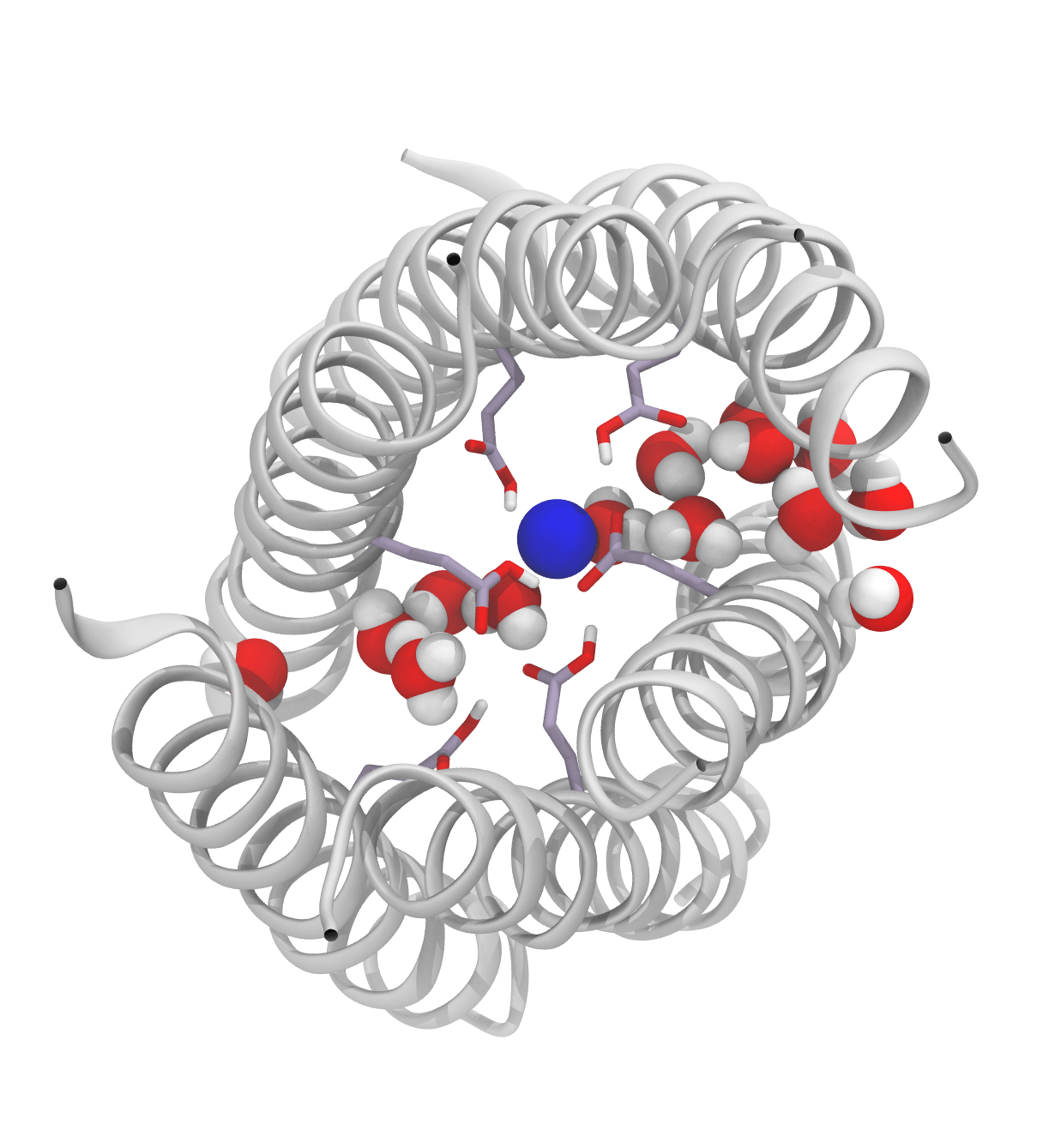
The optimum size of the QM region in QM/MM calculations is a matter of much debate in the literature. The effect of the size of the QM region was tested in solution (by adding additional water molecules to the QM region) and in the enzyme (by adding additional arginine side chains) showing that the difference in barrier heights predicted by common density functionals for any size of the QM region is significantly larger than the change in barrier caused by increasing the size of the QM region.
Ranaghan, K. E., Shchepanovska, D., Bennie, S. J., Lawan, N., Macrae, S. J., Zurek, J., Manby, F. R. and Mulholland, A. J.
J. Chem. Inf. Model., 2019
DOI: 10.1021/acs.jcim.8b00940