In a recent publication in PNAS, Corey et al (Collinson Group, School of Biochemistry, University of Bristol) work on the bacterial form of the general secretion system – aka the SecY machinery. This complex carries out the bulk of pre-protein secretion at the bacterial plasma membrane, powered by both ATP and the proton-motive force (PMF). They were interested in the interaction of SecY with the energetically-important cardiolipin (CL) molecule. CL is thought to be involved in many different bioenergetics processes, and has been previously implicated in SecY activity.
To approach this problem, they coupled the high predictive power of coarse-grained (CG) molecular dynamics (MD) with experimental analyses. Considerable speed up on atomistic simulation can be achieved using CG force fields, such as the Martini force field for biomolecules. By reducing the degrees of freedom of a system, it is possible to achieve sampling orders of magnitude faster than atomistic simulation – driven primarily by an increase in permissible MD step size, a reduction in interactions to compute per step and a smoothing of the energy landscape.
The CG data revealed two distinct CL binding sites on the SecY surface, which they were able to validate using native mass spectrometry (nMS), with Dr Argyris Politis at King’s College London, and FRET-based analysis on carefully designed variants of SecY.
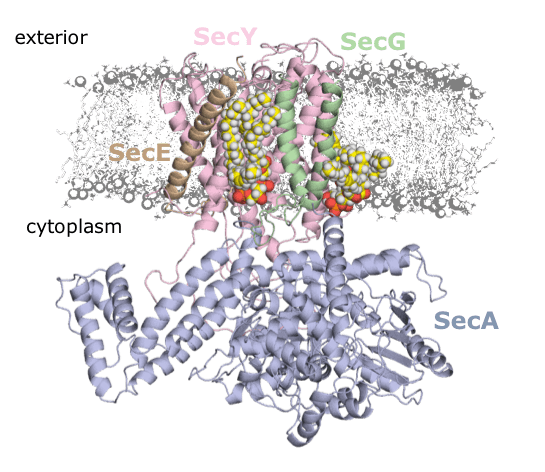
They then used these knockout variants to more deeply understand the importance of the SecY-CL interactions. Using a range of biochemical assays, they reveal that the specific interaction of CL at these sites is responsible for the previously-recorded heightened activity of SecY. Moreover, they establish a hitherto unknown role for CL in the PMF-driven activity of SecY. This is the first direct evidence of CL acting directly with the PMF in any bioenergetic system.
